To use the NextGENe Overlap Merger tool
1. On the NextGENe main menu, click Tools > Overlap Merger.
The Overlap Merger dialog box opens.
2. In the Input files pane, click Add to browse to and select the input files that are being merged.
3. In the Output field, you can leave the default value for the location of the output files as is (the default value is the directory path for the first data file added), or you can click Set to select a different location.
4. Specify your settings as appropriate.
Setting | Description |
|---|---|
Merge overlapping contigs | Applicable only for de novo assembly results. Determines whether any of the contigs are overlapping and can be merged further. |
Merge overlapping paired reads | Applicable only for raw paired reads that are overlapping.Keeps and merges the higher quality base of a pair. Note: The library size and read length determine whether the paired reads are overlapping or not. |
Merge cell-free paired ends | Discards low quality reads from the resultant output rather than merging them. |
Output reverse complement | Applicable only for merge cell-free paired ends. Generates the reverse complement for the sequenced reads. |
Overlap min bases | The minimum number of bases that must overlap for the contigs or paired reads to be merged. |
Ignore Low Quality Ends for Non-Overlapped Pairs | Applicable only for merge overlapping paired reads. Applicable only for elongated paired reads data. Non-overlapped reads are saved in the unmatched.fasta files. If elongated reads are used for merging, then lowercase letters, which are used at the ends of elongated reads, are trimmed from the non-overlapped reads before the file is saved. |
Merged Length [ ] bp to [1000] bp Merged Length [70] bp to [130] % of the longer read length | Applicable only for merge overlapping paired reads. Set an acceptable length for the merged results. Note: Both options can be selected. If both options are selected, then the data must meet both criteria to be included in the results. |
Trim Primer | Applicable only for merge cell-free paired ends. Trims reads where the specified sequence occurs, which removes primers. Note: If you select this option, then you must click Primer file, and then browse to and select the file that details the sequences that are to be trimmed. For detailed information about the format for this file, see Trim by sequences in the file. |
If you add multiple input files and you select Merge overlapping contigs, then both files are used for merging. For example, a contig from file A could be merged with a contig from file B. |
5. Click OK.
A folder is created for the output files. The default folder name is based on the name of the files that were analyzed and is appended with the word “Merge” as shown in the figure below.
NextGENe Overlap Merger output folder and files
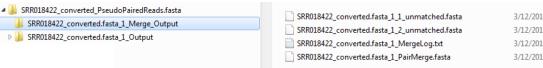
The folder contains several text files, which are detailed in the table below.
File | Description |
|---|---|
Merge Overlapping Contigs | |
input file name_ContigMerge.fasta | Contains the merged contigs. |
statinfo.txt | Details various statistics about the merge. |
Merge Overlapping Paired Reads | |
• File name 1_unmatched.fasta • File name 2_unmatched.fasta | Contain the reads that were not merged. |
MergeLog.txt | Details various statistics about the merge. |
PairMerge.fasta | Contains the merged reads. |