The NextGENe Format Conversion Tool
The NextGENe Format Conversion tool converts the format that the instrument uses to organize reads and assign quality scores to a standard .fasta format that NextGENe can read. In .fasta format, comment lines are marked with the greater than (>) symbol. The comment line contains the name that is assigned to a read. The sequence read base call line follows the comment line.
Example of a NextGENe .fasta file
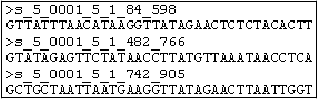
The figure above shows three of the reads in a .fasta file that is named is “s_5.fasta.” Each sequence read contains 36 nucleotides, and the name assigned to each read (from top to bottom, respectively) is: _0001_5_1_84_598, _0001_5_1_432_766, and _0001_5_1_742_905. You can specify values for quality settings to trim or remove low quality reads before you convert a supplier’s format to NextGENe’s .fasta format.