Genomic regions or genomes smaller than 250 Mbp
For genomic regions or genomes smaller than 250 Mbp, NextGENe uses an alignment method that is similar to BLAT methodology to align sequence reads to the reference. The reference file is first divided into an index table. Every 12 bases of each sequence read is aligned to this table. The positions of alignment between the reads and the reference are determined and the alignment is evaluated linearly. If they are in a line, the sample sequence can be aligned to the reference target positions. (Jumps might exist in the line because of true or false positive indels.) Reads can be matched to a single position, or they can be matched to multiple positions. If a read matches exactly at more than one position, it can be aligned at each exact match position when “Allow Ambiguous” is selected. (See Allow Ambiguous Mapping.) If this option is set equal to one, the read is aligned to the first exact match position from the beginning of the reference. If this option is set equal to zero, all reads that match perfectly at more than one location are discarded.
The Allow Ambiguous setting is not applicable for reads that include mismatches. Instead, when reads match to more than one position with the same number of mismatches, the Uniqueness score is used to determine the best position to which to align the read. The uniqueness score is calculated according to the following, where “n” is the number of hits on the reference:
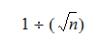
The region with the greatest Uniqueness score is selected to align the read.