STR Expansion Analysis
NextGENe®LR software can be used for STR Expansion analysis from long read sequencing data such as< data from Pacific BioSciences systems. This provides an accurate reporting of STR lengths to identify STR loci with lengths associated with disease.
NextGENeLR aligns the long sequence reads to the reference sequence using a specialized alignment algorithm. Alignment results are saved to a BAM file which can be loaded in the built-in Alignment Viewer for visualization and reporting. When the STR Expansion Report is opened, the STR regions to be evaluated are defined. A default list of STR regions associated with disease is provided. This list of regions can be modified by removing regions, adding regions, editing regions, or uploading a text file containing a list of regions to be added. For the STR regions of interest, the number of reads with each repeat length is counted.
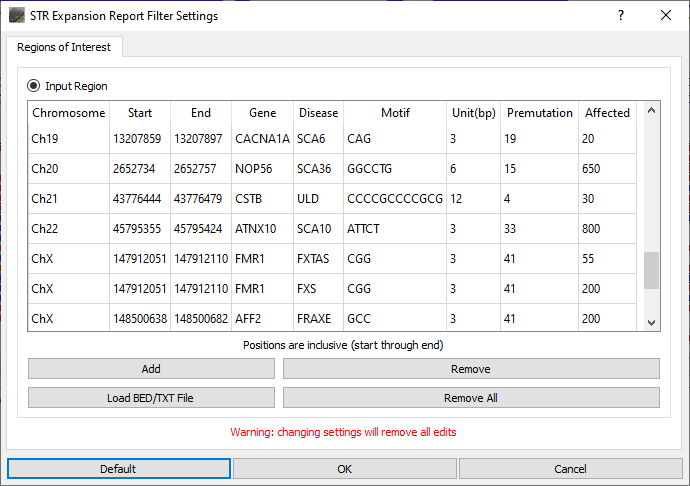
Figure 1: STR Expansion Regions of Interest can be defined. The default list can be used as is, regions can be removed and/or added, or a custom list of regions can be loaded.
Repeat lengths for all included regions of interest are displayed in the graphical STR Expansion Report. The normal range for the repeat length is displayed with green. Gray denotes the premutation range and red indicates the repeat length associated with disease. Blue boxes are placed at the repeat length(s) detected for the sample. The top number for each blue box indicates the repeat length and the bottom number indicates the number of reads with this length.
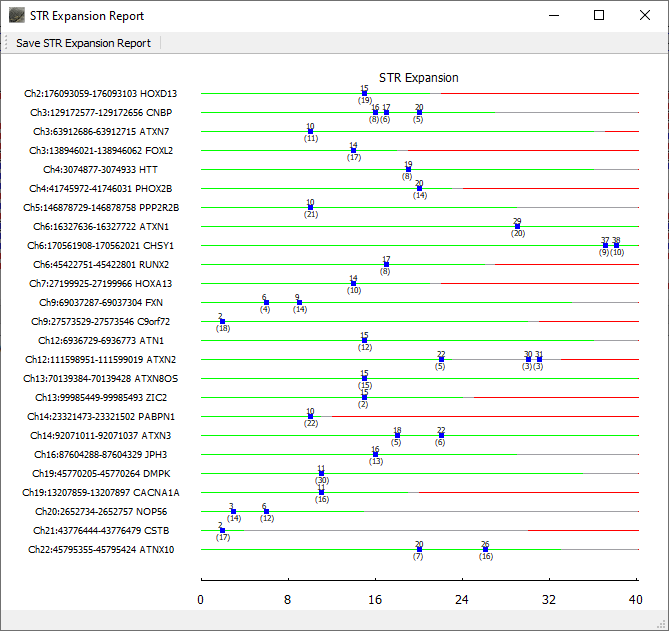
Figure 2: The NextGENeLR STR Expansion Report displays the repeat lengths detected for all STR regions of interest. Green indicates the normal STR length range, gray for premutation, and red for affected. In this case, the ATXN2 STR region shows some reads in the premutation range.
Application Notes:
Trademarks property of their respective owner