NEBNext Direct Analysis using Unique Molecular Identifiers (UMIs)
Processing NEBNext Direct data is facilitated with the use of NextGENe software's AutoRun Tool. The AutoRun Tool includes built-in templates for single click processing NEBNext Direct data.
The templates include removal of PCR duplicates based on UMIs. The usage of UMIs for duplicate removal allows for increased allele frequency accuracy to improve the accuracy of variant detection. The NEBNext AutoRun templates can be used to quickly set up a batch analysis for multiple samples with just a few clicks.
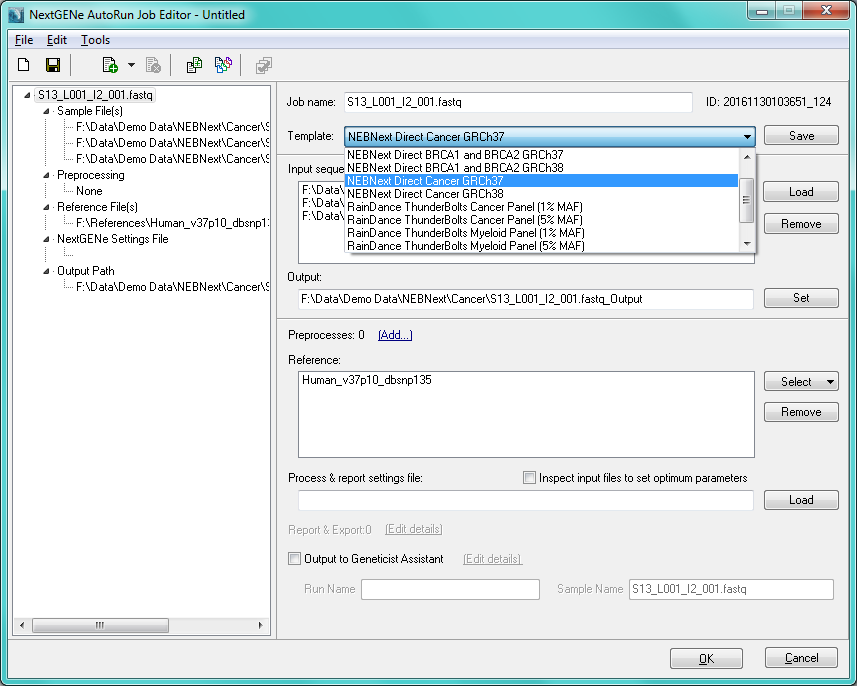
Figure 1: NextGENe AutoRun Tool with NEBNext Direct templates
The NextGENe NEBNext Direct Template automatically guides the sample files through a series of steps, including Removal of PCR duplicates, Format Conversion, trimming of adapters, alignment to the human genome and variant calling. Variants will be called when all of the following criteria are satisfied:
-
Percentage of reads with variant is greater than 1.5% -
Variant is found in more than 3 reads -
Total coverage is more than 100 reads -
Variant forward/reverse balance ratio more than 0.2 (0.8 for homopolymer indels) -
Variant is within target region
NextGENe software uses UMIs within the Illumina I2 files to identify PCR duplicates. Within a UMI family, the pair of duplicates for each template with the highest total score is maintained to be processed along with unique paired reads, while duplicate reads are removed from further processing.
Without duplicate removal, many reads for a region appear identical, starting and ending at the same positions. After duplicate removal, a more tiled read alignment can be seen. Some reads that appear as duplicates may still be seen (Figure 2). Since duplicate removal is performed based on the I2 UMI sequences, only true PCR duplicates are removed.

Figure 2: Read pile-up view wthout duplicate removal (left) and with duplicates removed (right)
After processing, the projects can be visualized in the NextGENe Viewer (Figure 3). Several reports, including the mutation report, several coverage curve reports (reporting low coverage regions using different coverage cutoffs), and an expression report (showing read counts per target) are automatically exported based on the template settings. New reports can also be created and saved through the NextGENe Viewer.
The NextGENe Reference and Track Manager tool allows for optional tracks to be imported into NextGENe software. These tracks, including ClinVar, gnomAD, and dbNSFP, can then be queried automatically for every new project, giving additional information for each variant to be included in the Mutation Report.
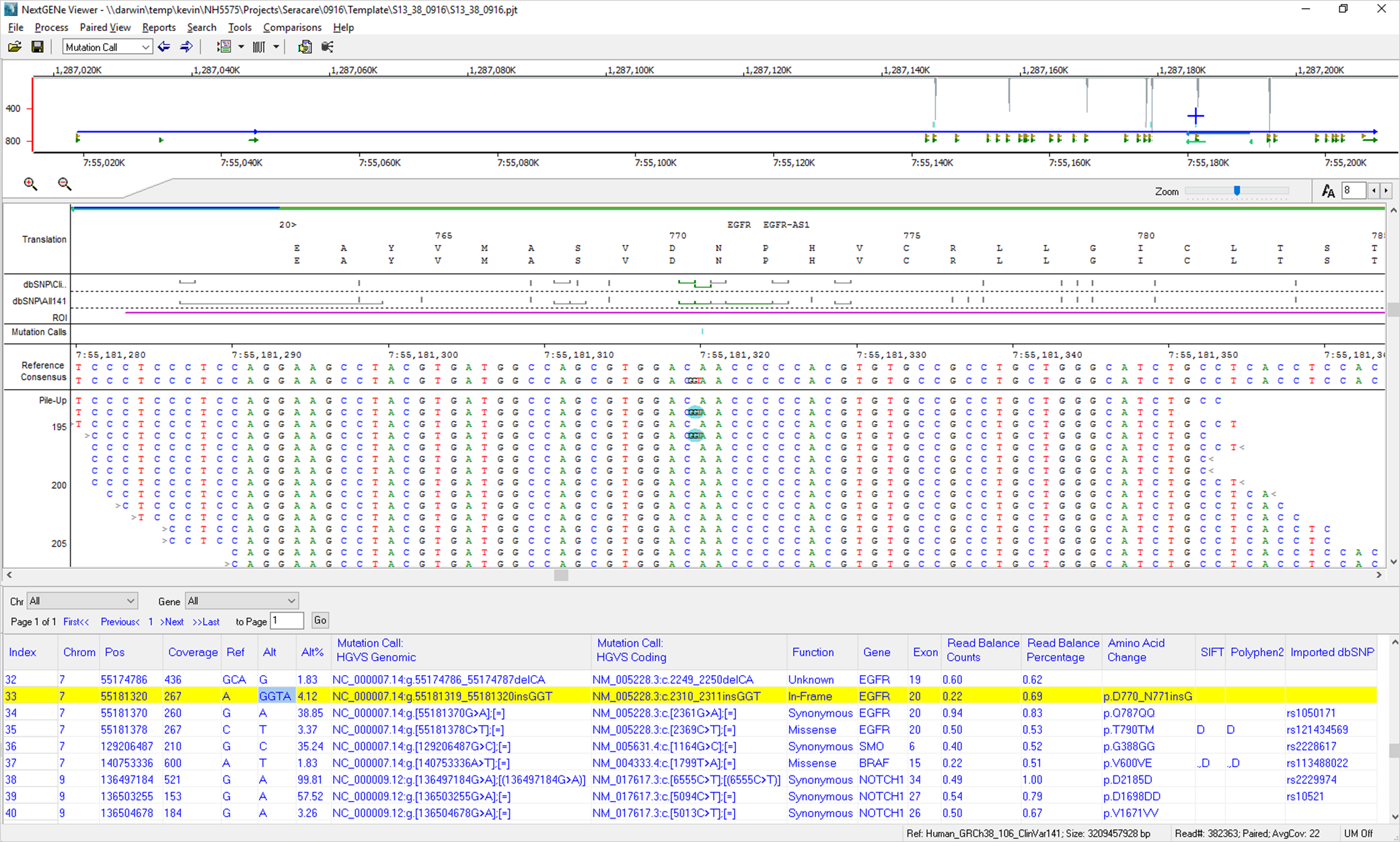
Figure 3: NextGENe Viewer
Application Notes:
Webinars:
- Creating Sequence Alignment Projects with NextGENe
- Creating Projects with the NextGENe Software AutoRun Tool
Pricing & Trial Version:
Reference Material:
Trademarks property of their respective owner