Insertion and Deletion (Indel) Detection from Sanger Sequencing Traces
Mutation Surveyor software employs a rigorous, multi-step alignment algorithm to detect insertions, duplications and deletions from Sanger Sequencing traces. Compatible with outputs from all major DNA capillary electrophoresis platforms and sequence file types (.ABI, .AB1, .SCF), Mutation Surveyor software’s unique alignment algorithm accurately detects both homozygous and heterozygous insertions and deletions by monitoring sample trace and reference trace mobility. Mutation Surveyor software is capable of aligning sequence traces containing up to 20% variation from the reference, and features the ability to detect small indel events as well as large indel events up to 15% of the sequencing trace. Included in the package is a unique Heterozygous indel Detection tool, which automatically deconvolutes the heterozygous mutant allele from the wild type allele. Along with its patented “anti-correlation” technology (U.S. Patent 8,086,410), the enhanced alignment and detection of Mutation Surveyor software provides enhanced accuracy and sensitivity over similar programs such as Thermo Fisher Scientific SeqScape®, Variant Reporter™, and Minor Variant Finder; DNAStar's SeqMan Pro™; Gene Code's Sequencher®; or JSI Medical Systems' SEQUENCE PILOT.
Enhanced Detection and Visualization of Insertion, Duplication, and Deletion Events
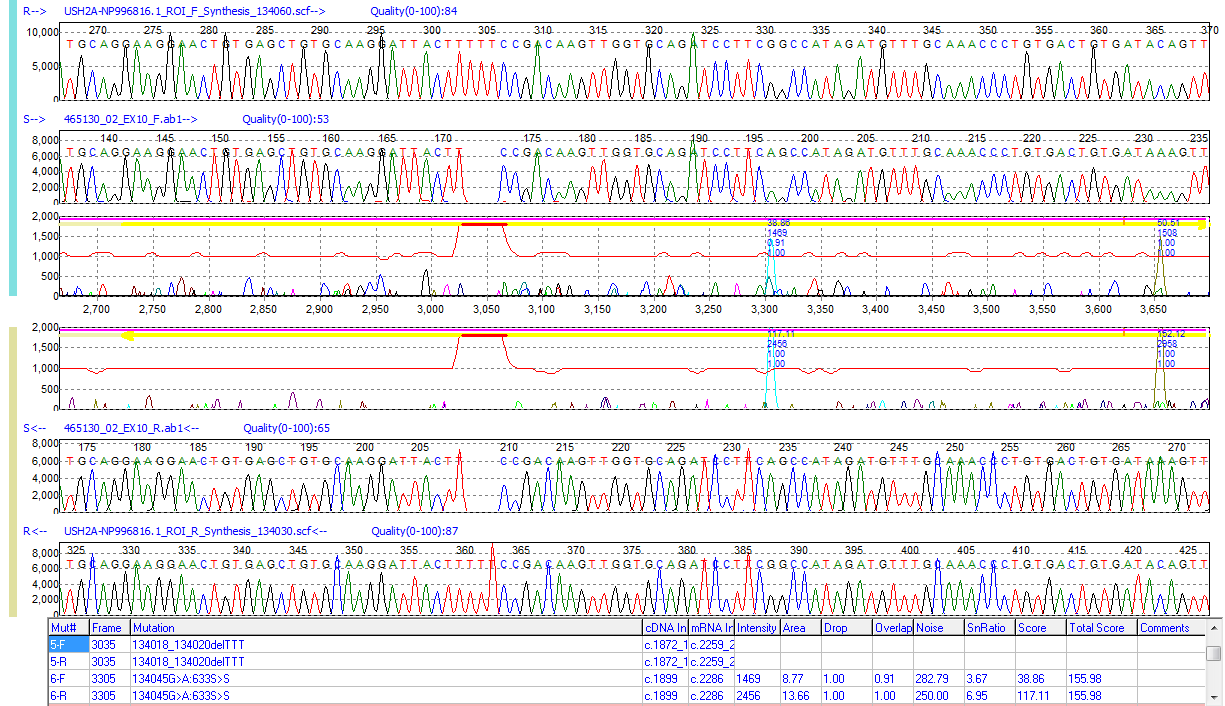
Figure 1: A 3bp homozygous deletion detected in a sample trace. The sample trace is gapped to continue alignment to the reference and a red bar is placed in the mutation electropherogram at the location of the event. The software uses the mobility line, which runs horizontally along the middle of the Mutation Electropherogram, to indicate differences in mobility between the sample and reference.
Mutation Surveyor software offers enhanced detection, visualization, and reporting of insertions and deletions, making it easy for the researcher to identify and edit variants. The software denotes differences between sample and reference mobility through changes in the mobility line in the Mutation Electropherogram, providing an extremely low false positive rate and ignoring miscalls or overcalls from basecall software. When an indel event is detected, a color change from green to red in the mobility line indicates to researchers the presence of a homozygous indel event. The mobility line will also display a negative (insertion) or positive (deletion) spike as well as a thick, red bar over the location of the event. Heterozygous events are also displayed in a similar manner, with brown bars indicating the location of heterozygous insertions or deletions.
Robust Multi-Step Sequence Alignment Enables Detection of Longer Indels up to 15% of Sample Trace
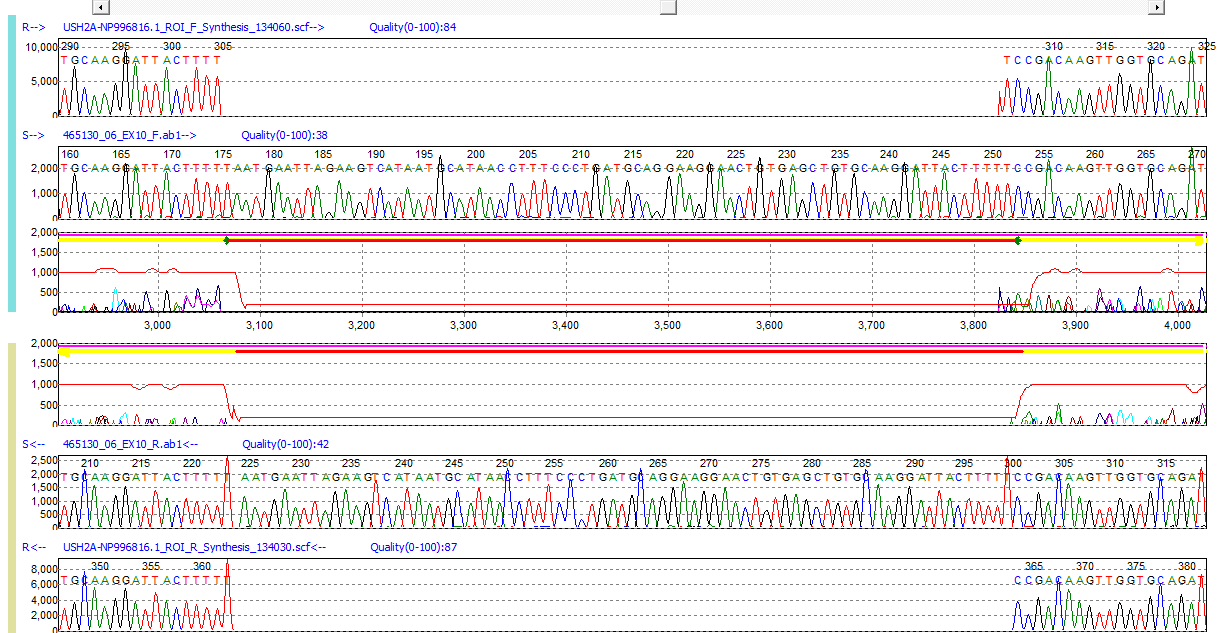
Figure 2: A 76bp homozygous duplication detected in a sample trace. While monitoring the mobility of the sample and reference, Mutation Surveyor software gaps the reference to detect the insertion event and continues alignment. This insertion accounts for 15% of the sample trace.
Mutation Surveyor software utilizes a multi-step alignment algorithm which is capable of aligning sequence traces containing up to 20% variation from the reference as well as indels up to 15% the length of the sample sequence trace. The first step of the alignment process utilizes a 12bp seed to align samples to the reference trace. If a gap occurs within the 12bp seed, the algorithm reduces the size to 3bp in the local region, followed by the Smith-Waterman algorithm to complete local regional alignment. In this way, the software is able to detect both large and small indel events. As realignment proceeds the software automatically gaps the reference trace (insertion/duplication) or sample trace (deletion).
Heterozygous Indel Detection and Deconvolution
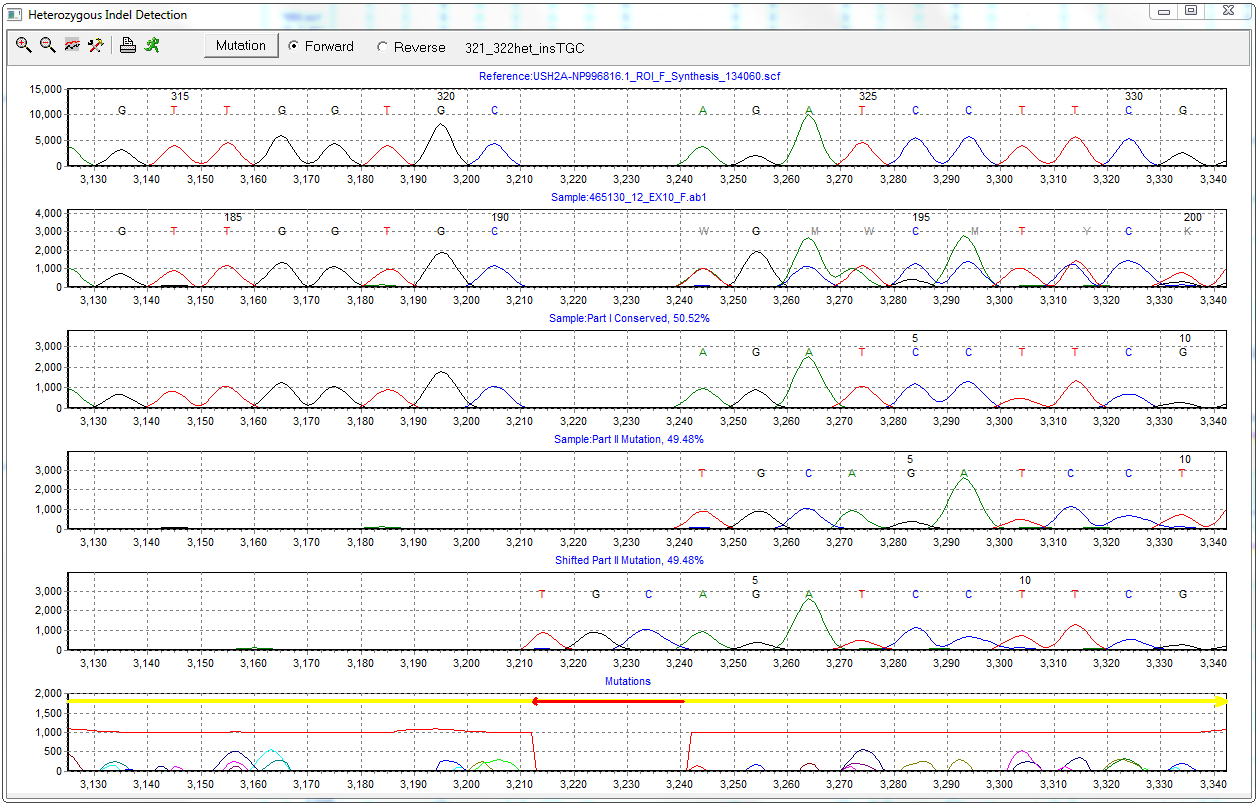
Figure 3: The Heterozygous Indel Detection tool detected a 3bp heterozygous insertion of TGC and was able to deconvolute the sample into normal and mutant alleles.
In the event that a heterozygous insertion or deletion is detected, the software will automatically deconvolute the sample into two clean traces: wild-type/normal allele and novel/mutant allele. The mutant trace is then shifted to align to the reference, indicating the position and nucleotide sequence of the indel event. For more information, please see the Indel Deconvolution page.
Trademarks property of their respective owner