Mitochondrial DNA Sequence Analysis
Mitochondrial DNA sequencing has become a useful tool in biological and medical disciplines, including the study of diabetes, cancer, and forensic identification. These studies often require sequencing projects of the mitochondrial genome that cover both coding regions and non-coding regions, such as the hypervariable regions located in the D-loop. However, the circular nature of the genome and differences in the mitochondrial amino acid codons present a challenge for precise variant calling in mtDNA sequences. Mutation Surveyor software offers solutions to these issues while providing high accuracy and sensitivity in the detection of single nucleotide polymorphisms (SNPs), homozygous and heterozygous insertions and deletions (indels), and low-frequency variants such as heteroplasmy. Along with accuracy greater than 99% in the bi-directional analysis mode and sensitivity to 5% of the primary peak, Mutation Surveyor software automatically aligns sequences to the revised Cambridge Reference Sequence (rCRS), correctly spans the origin (D-loop) of the mitochondrial genome, and accounts for the amino acid differences relative to the vertebrate mitochondrial genetic code.
Sequencing Across the Region
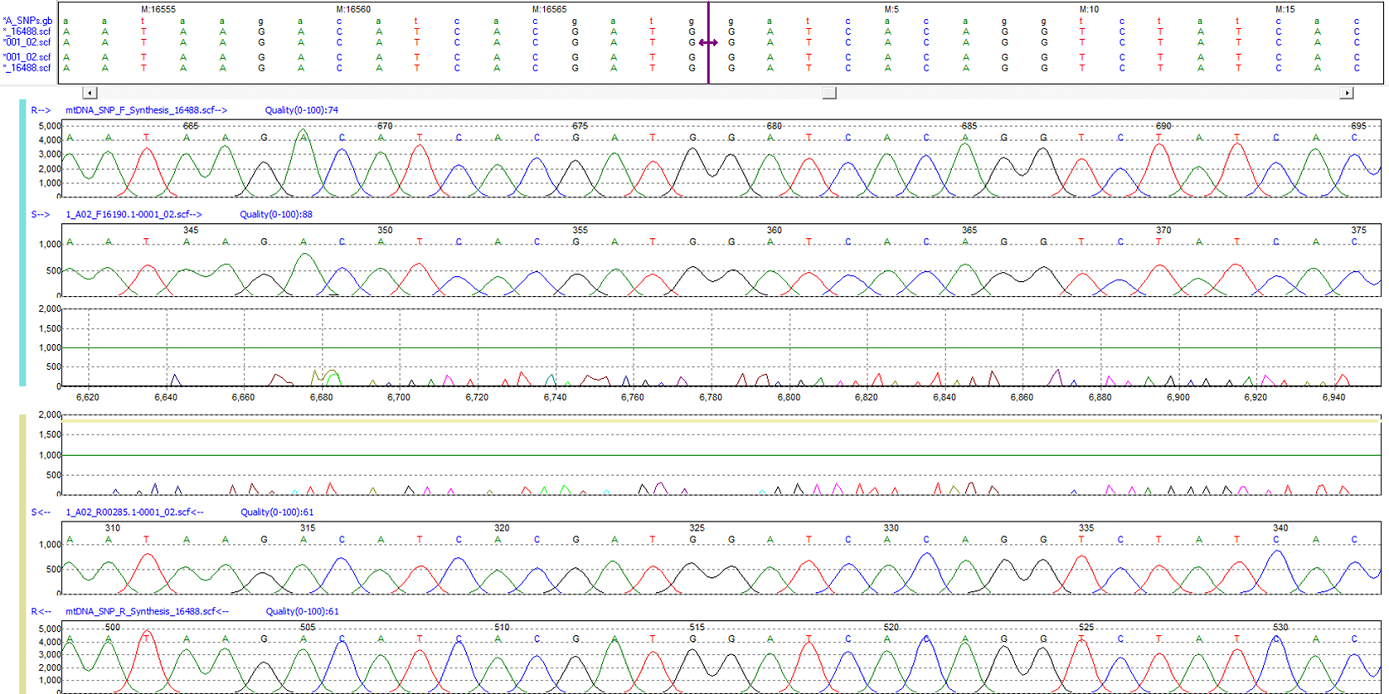
Figure 1: The purple bar in the nucleotide sequence frame represents the origin of the mtDNA genome.
The hypervariable regions of the mitochondrial genome are separated by an area within the control region, which marks the ending and beginning of the genome annotation. Mutation Surveyor software automatically recognizes a tag in the mtDNA GenBank file, enabling the program to identify the origin of the D-loop separating the HV1 and HV2 regions. When sequences spanning the origin are loaded into Mutation Surveyor, nucleotides on both sides of the origin are aligned, and the software places a purple bar in the nucleotide sequence frame of the Graphical Analysis Display (GAD) to display the origin between position 16569 and position 1.
Annotation of Mitochondrial Genetic Code
Mutation Surveyor software Recognizes Amino Acid Translation from Different Genetic Codes
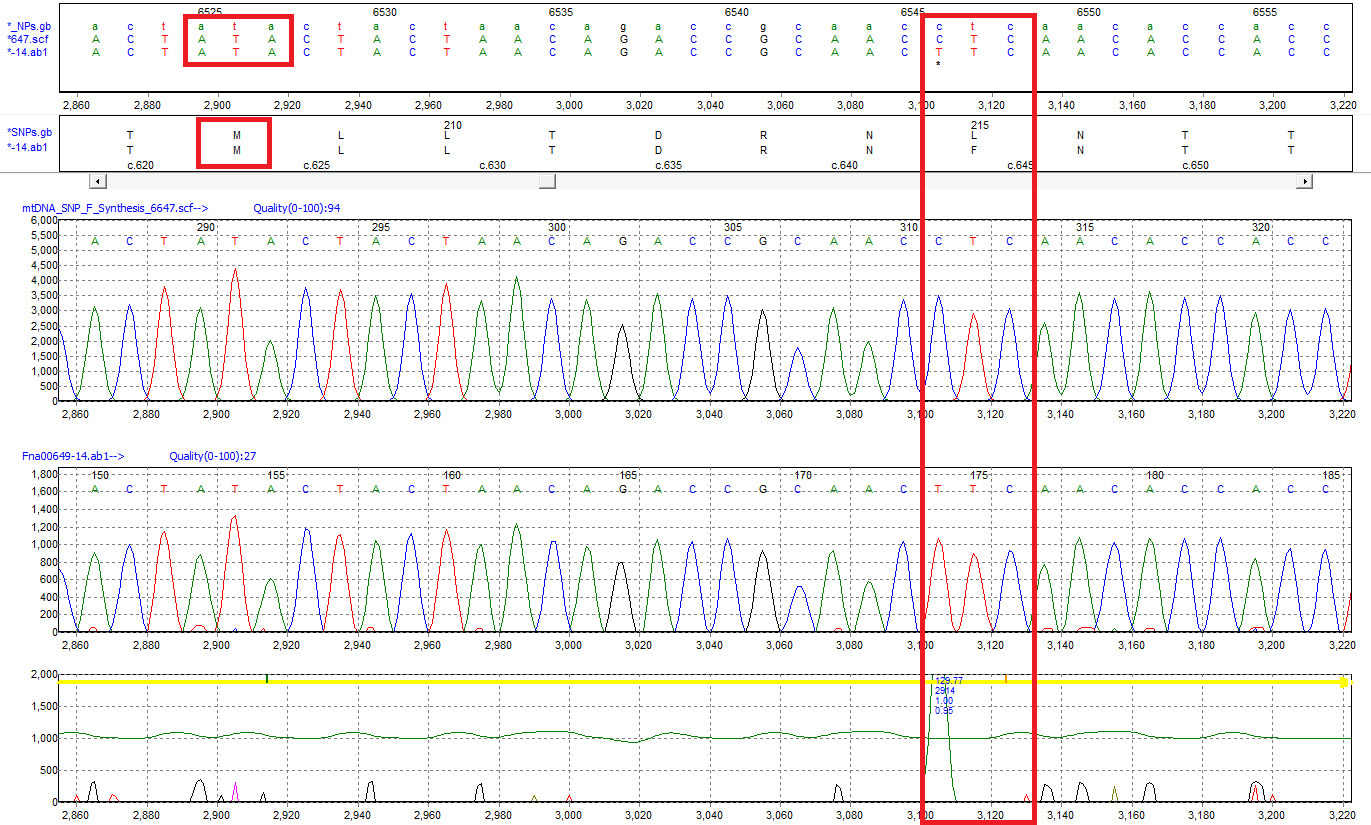
Figure 2: The ATA codon translated to Isoleucine in the standard genetic code is read as Methionine using the mtDNA GenBank file. A SNP of C>T is detected at position 6546 and results in an amino acid change from Leucine (L) to Phenylalanine (F).
Mutation Surveyor software annotates the sample sequences according to the mitochondrial genetic code without any user intervention by reading the translation table within the GenBank file. In this example, the "ATA" codon that codes for Isoleucine (I) in the standard genetic code is translated as Methionine (M) using the vertebrate mitochondrial GenBank file. Amino acid changes caused by detected variants are also annotated relative to the mtDNA code, and variants are annotated by their position in the rCRS.
Robust Sequence Alignment and Accurate Variant Identification
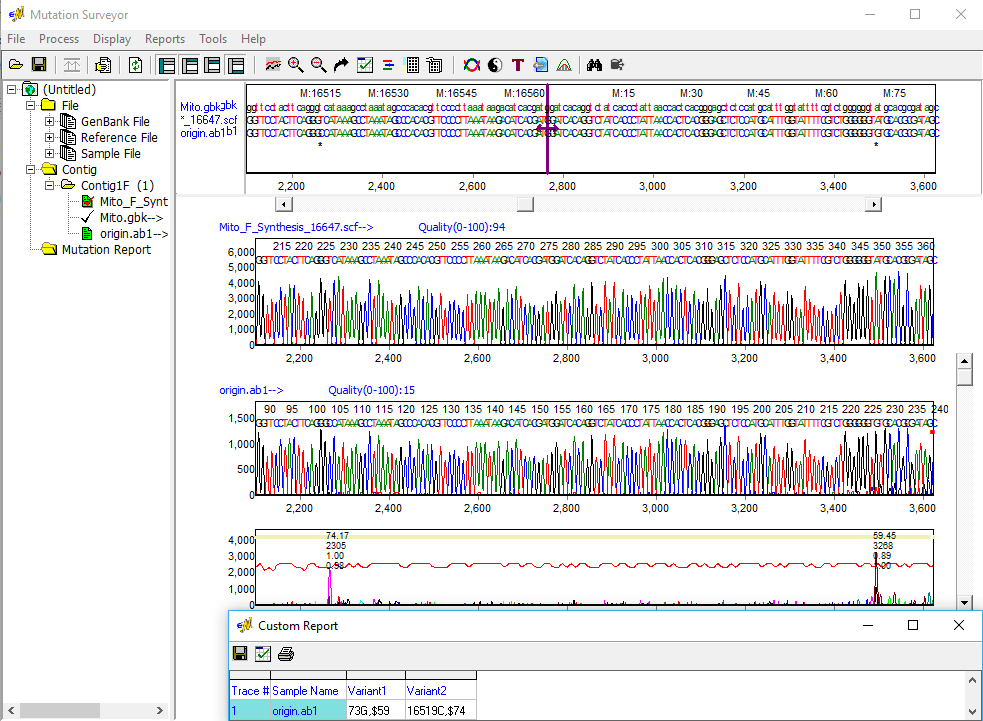
Figure 3: Variations are detected on both sides of the origin in a single sequence. Mutation Surveyor software accurately aligns highly variable sequences and detects variations with greater than 99% accuracy, and the Custom Report displays mutation call nomenclature following SWGDAM guidelines.
Mutation Surveyor software is able to quickly and accurately align mtDNA sequence traces to the revised Cambridge Reference Sequence (rCRS), including hypervariable sequences spanning the HV1 and HV2 regions for identification of variants associated with human mitochondrial DNA halplogroups. Variants are detected using the same "anti-correlation" technology used with genomic DNA sequences, providing an increased level of accuracy and sensitivity not found in other software. The Custom Report can display calls following the SWGDAM guidelines, such as 152G, 309.1C, and 522d for substitution, insertion, and deletion, respectively.
Application Notes:
Webinars:
Trademarks property of their respective owner